
作者: 东华帝君


新药研发,是一个涵盖多学科共同努力、富集多部门共同协作,而“梦想”完成的系统工程。但由于研发过程的高风险特征,故纵使每个环节都倾注其所有,最终亦未必能开花结果。在这里,如果将立项比作“撒种子”,那IND过程则犹如“开花”,NDA过程就类似于“结果”。项目在立项之初,撒下千万颗种子,最终能长出果实的,绝对凤毛麟角。而在现实工作中,如能真正成长到开花阶段的IND,就已经让人超级敬佩、超级羡慕了。因为,在这一过程中,深处项目当中的研发人员,大部分均已竭尽全力,且得到了幸运女神的眷顾。
IND简介
IND,Investigational New Drug,一般是指尚未经过上市审批,正在进行各阶段临床试验的新药,实际应用中,IND或CTA (clinical trial application)已变成药品上市前人体临床试验的代名词。IND申请可能是一个,也可能是序贯的一组研究,目的在于获得产品安全性和有效性的证据。一般来说,IND可分为商业IND和非商业IND,商业IND是以注册为目的,可区分I期临床试验、II期临床试验和III期临床试验,或探索性临床试验和确证性临床试验。商业IND通常由药物研发申请人发起,非商业IND通常不以注册为目的,多由研究者发起,难以界定应归属于探索性临床试验或者确证性临床试验。
IND申报处于新药研发的哪个环节?
创新药物从结构设计到发现、再到最终的获批上市,经历的是一个充满荆棘的系统的炼狱过程。简要的说,首先是新化学实体NCE的发现,其主要包括先导化合物、候选药物的确定;之后进入到临床前研究阶段,这一过程主要包括药理、毒理、药代、处方前研究等内容;待完成初步临床前评价后,即将进入临床研究之时,开发公司申请人须向相应药审部门进行注册申报,这个节点就是本文要讲的内容~IND申请;待药审部门通过IND申请或者没有反馈意见时,项目便进入到下一阶段,即I、II、III期临床研究;待临床前研究、临床研究全部或部分完成之后,如果达到了预期目的,即可提交新药上市申请NDA,以求获批上市销售;上市后,开发公司仍需对产品进行IV期临床研究和上市后监测,使之更加充分的理解药物的机理、范围、治疗作用、副作用,等等等等….
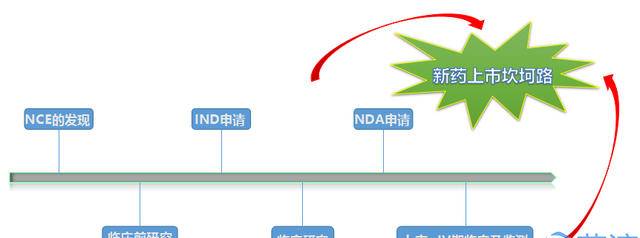
再简要概述IND申报过程
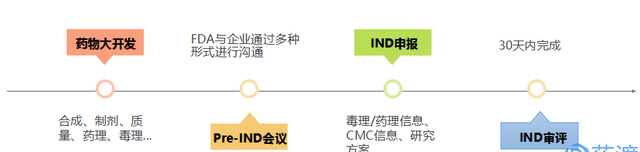
如上所述,开发公司提交IND申请之前,需要对药物进行一系列的临床前研究,从而为临床研究铺垫强有力的安全性、有效性数据。当收集足够的数据之后,便可形成报告,开始准备IND申请文件。这里建议下,IND申请准备的时间一定要足够充分,最好在确定候选药物之后,便准备IND申请文件,FDA官网有许多IND相关的指导文件和表格(https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/investigationalnewdrugindapplication/default.htm),可以在项目跟进的同时,加以学习,从而使申请文件内容清晰合理。
在申请人递交IND申请前,可自愿与FDA进行一次重要的会议,即Pre-IND会议,期限为申请日开始后60天内。该会议上,申请人可以同FDA相关专家共同讨论,从而确定试验方案、讨论I期临床试验设计范围、新药儿科研究计划、现有CMC信息是否充分等等。当然,并非所有的IND都需要召开该会议,一些较简单的IND申请可不必召开,但对于那些严重危害生命的疾病、新适应症等,最好还是共同讨论一下比较保险……
上述过程准备充分之后,即可向药审部门提交IND。在美国,FDA的项目监管人员会将IND申请转交给由化学家、药理学家/毒理学家、临床医生、统计学家和药代动力学家组成的审查小组。FDA也会在30天内,对IND的安全性进行审查,从而确保受试者在试验过程中的安全保障。在此期间,FDA可能会要求申报公司提供额外的信息和说明,故申请人最好提前进行充分且适当的数据储备。如果30天内,FDA没有任何的信息反馈,企业即可认定IND审核通过,按照申请内容开展相应的临床试验。
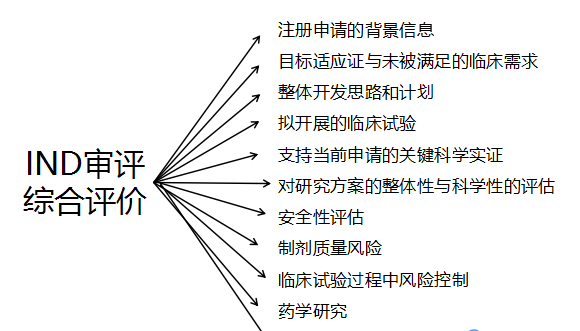
IND审评都会评价哪些信息?
☆注册申请的背景信息
IND申请需考虑的背景信息主要包括4个部分:即化合物的结构来源、药理学分类、国内外同类药物研究/上市现状、申请项目的基本状况。其中,结构来源主要分析化合物是全新结构、已有母核结构改造或已上市药物的成盐或成酯等;药理学分类主要结合申请人提供的研究信息判断其为新机制新靶点还是已有成药的作用机制或靶点。以上两点,即可对其成药性与风险性作初步判断,例如全新结构、基于新机制新靶点的化合物,其所隐含的开发风险远高于已上市药物的成盐或成酯,其成药的可能性也相对较低。
☆目标适应证与未被满足的临床需求
一方面可能是为了增加药物的可及性,一方面可能是为了解决现有药物存在的问题。因此需要充分了解目标适应证,包括流行病学证据、疾病的严重程度和预后情况、目前已有治疗手段及其不足、目标适应证药物研发趋势与经验等。此外,还需了解相关诊疗指南、药物研发趋势及在申品种的潜力等等。
☆整体开发思路和计划
主要关注申请人是否已制定人体试验研究路径的整体规划,所申请的研究处于规划中的哪个阶段,或尚未有整体或阶段性研究规划,仅仅就申请项目本身提出了特定阶段的研究问题/方案。
☆拟开展的临床试验
了解申请项目的基本情况,如拟开展哪一期/哪一阶段的人体临床试验,其中包括受试样本量、试验持续时间、给药方案、研究规模(地点)等,为后续技术评价提供参考基础。
☆支持当前申请的关键科学实证
关注支持当前申请的依据,主要是从有效性立题依据方面考虑,这些支持性的证据主要来源于临床试验(在某个化合物的临床试验过程中,可能会发现新的临床应用,而新的应用往往与原来预期的适应证相差甚远)、基础研究(目前从实验室开始的基础研究,仍是大多数药物开发的标准路径,如果支持性信息来源于基础研究,需关注这些信息的证据强度)或其他相关信息(有些情况下,特别是已有同作用机制的药物成功上市、没有合理的模型时,可通过桥接性信息获取对申请项目的支持性信息,如作用机制研究、药物代谢分布研究信息等)。
☆对研究方案的整体性与科学性的评估
从目前国际研究趋势来看,新药开发是以临床试验方案为主导的研究评价模式。传统的临床试验分期越来越模糊,一个药物能否上市取决于获得的数据能否支持其安全性、有效性的评价,而非是否进行了完善的各期临床试验。非临床及药学研究的内容和时间安排要根据拟定进行的临床试验内容来确定,而研究与评价是伴随而行的,边研究边评价边决策。对研究方案的整体性与科学性的评估主要是对拟定的临床方案进行评估,涉及内容包括基本研究假设、研究人群、疗效指标、研究周期、给药方法和依据、控制偏倚手段、数据管理、申请人的风险管控措施等。
☆安全性评估
基于拟定的临床试验方案,对化合物自身研究数据、同类化合物或同类作用机制药物的研究数据以及制剂相关的安全性信息进行评估,判断现有数据(自身+文献)能否支持临床试验的安全性,并提出可能需要注意的风险控制点。而风险主要来源于化合物自身、同类化合物、同类机制等研究。
☆制剂质量风险
用于早期临床试验的受试物质量与非临床试验的受试物质量、后续临床试验采用的受试物质量与前期已完成临床试验采用的受试物质量是否具有可比性,关注受试物所含杂质的性质与含量。
☆临床试验过程中风险控制
基于上述对拟定的临床试验方案、安全性信息的评估,分析临床试验方案中是否有足够的风险控制措施。具体内容包括:临床试验设计中受试者是健康志愿者还是根据安全性风险信号和临床试验本身选择相应的患者群;针对已知潜在风险,监测程序是否完善,能否有效暴露并能识别出安全性信号,是否有不能接受的系统风险;临床试验方案是否基于风险分析制定了必要的风险控制手段;是否有针对风险控制手段评估改进的机制。
☆药学研究
在创新药研发进程中,与药学相关的变更几乎是不可避免的,尤其是在早期开发阶段,变更发生得较为频繁。通常,在临床前和早期临床研究阶段,药学研究主要是为以上研究提供质量有保证的研究用样品;随着临床研究的推进,药学研究则致力于确定、稳定、重现、可工业化的生产工艺以及构建完善的药品质量控制体系;结合临床研究/治疗需要、放大生产需要以及对药物认识的不断加深,对剂型、规格、处方工艺、分析方法、质量标准等进行调整优化。
☆技术结论
归纳总结前述背景性信息和评价性信息,判断当前获取的证据是否支持申请人拟定的研究方案。如果支持,则需要明确支持的临床探索范围、相应的风险提示、后续需要进一步开展的支持性研究及其他相关方面如PI、CRO、伦理委员会的工作等。如果现有证据不支持拟定的临床试验方案,则需要明确其缺陷所在及后续应关注的问题。
让人沮丧的IND失败率
众所周知,在整个IND过程中都存在失败的可能。据报道称,IND总体失败率超过90 %,其中,I期临床试验中失败率在95%以上,II期和III期分别约为60%和25%。IND失败的原因是多方面的,1991年和2000年,10家大型制药公司的数据显示,研发的药物由于临床安全性因素被淘汰的案例比率分别约为10%和12%,由于临床有效性被淘汰的案例分别约为30%和28%,由于药代动力学不理想或者生物等效性因素被淘汰的案例约为40%和8%,由于商业因素被淘汰的案例约为5%和21%,由于非临床毒性被淘汰的案例约为11%和21%,由于其他不确定因素被淘汰的案例约为4%和8%。此外,2000年的数据显示,由于处方因素和价格因素被淘汰的比率分别约为5%和9%。从上述原因分析,导致药物研发失败的前3位因素分别是药代动力学、临床有效性和毒性。从分期来看,因临床有效性不够被淘汰的比率在II期和III期临床试验中相当,基本都在50%~55%。因安全性被淘汰的比率在I期和II期临床试验中相当,约为30%。
展望
对于我国来说,本土上市的1类新药并不多,再进一步讲,类似于国外大型制药公司的真正意义上的1类新药甚至还没有。但当下,国内的大环境又在迫切要求药物研发要尽快赶上欧美等发达国家,故国内创新药的春天,似乎真的来了。不过,如今创新药的开发已不同往昔,其难度越来越高,且国内新药研发的软硬实力,还不足以赶上当前对真正意义上创新药物的要求。故每当发现一份相对重量级的IND申请,大家都会对其关注颇多,希望其能够结出高质量的果实,改善国内创新药的大环境,同时也增加新药研发人员的信心。创新药物研发,绝对算得上是脑力、体力均极度疲惫的工作,但这份工作同样也激发着每一个新药研发人员与疾病作斗争的无限动力,希望每一个新药研发人员都能耐得住这份寂寞,伴随着一件件IND的申请,成长为真正配得上新药研发岗位的科研人员,当然,如果能进一步拿到一件NDA,那就更加完美了!互勉!
本网页内容旨在传播知识,若有侵权等问题请及时与本网联系,我们将在第一时间删除处理。E-MAIL:dandanxi6@qq.com